Our research focuses on dissecting general principles of genome regulation, including how the cis-regulatory genome is organised within the nucleus, and how chromatin state and transcription factors influence this process. Our work combines (single-cell) genomic, genetic, imaging and computational approaches to understand these processes, including the development of new genomic methods within the context of a multicellular embryo.
Genome Regulation & Enhancer Function
Expression states are initiated through diverse cues modulating transcription factor activity, which converge on regulatory elements in the non-coding genome called enhancers. Enhancers act as integration platforms, being bound by multiple transcription factors and co-factors, and they work together with structural elements to control specific patterns of expression, telling genes when and where to be expressed. Given their central role, mutations in enhancers often lead to devastating developmental defects and disease.
We are studying enhancer function during key transitions in embryonic development, dissecting the interplay between transcription factor (TF) occupancy, chromatin state and enhancer output. Equally important, we are also studying how multiple enhancers and other cis-regulatory elements work together to regulate gene expression. In addition to CRISPR deletions, we are using optogenetic methods we developed to very rapidly deplete transcription factors from the nucleus to uncover their direct regulatory input (Figure 1). iLEXY triggers the translocation of proteins from the nucleus to the cytoplasm within minutes, and is highly reversible (Kögler et al. Dev Cell 2022). Future directions will be to combine this with single cell methods and synthetic genomics to understand the regulatory properties of enhancers.
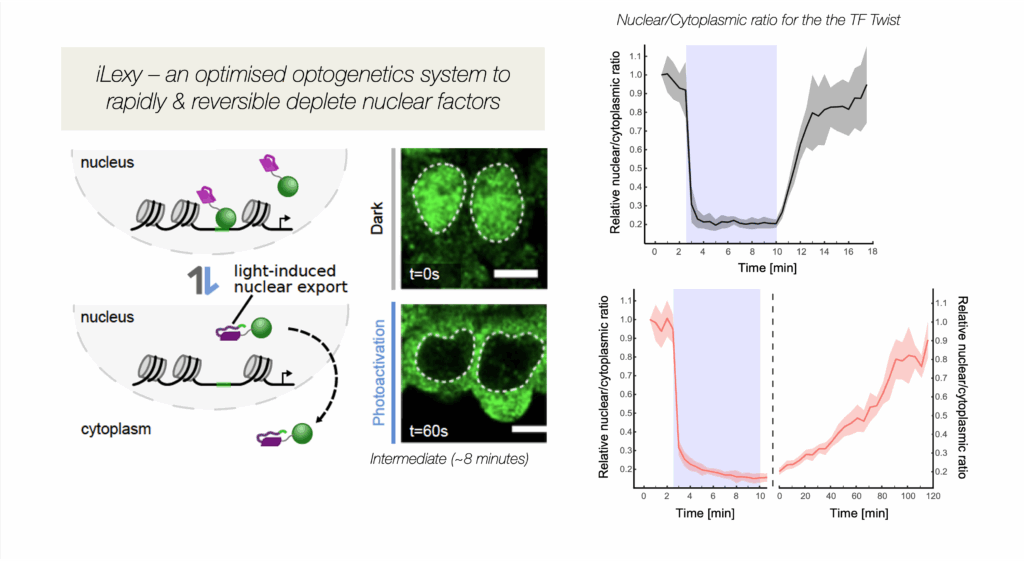
Figure 1. iLEXY, an optogenetic method to rapidly deplete transcription factors from the nucleus in embryos, in minutes: Shown in green is the transcription factor Twist in the nucleus in Dark conditions (safe light) and after 1 minute blue light exposure – the quantification is shown on the right. By mutating the LOV domain, we created versions with different strengths and recovery times (Kögler et al. Dev Cell 2021).
3D Chromatin Topology
To fit into the nucleus, the genome is tightly folded at multiple scales, yet this packing must allow for gene regulation to occur. For example, for enhancers to function, they must come into physical proximity to their target genes. We previously found that at early and mid-stages of embryogenesis, developmental enhancers are often in proximity to their promoters hours before gene expression (Ghavi-Helm et al. Nature 2014). These preformed topologies often contain paused polymerase at the genes’ promoters, and may therefore be primed and ready for very rapid activation of transcription. However, this changes at later stages during tissue differentiation, where many new, more long-range tissue-specific enhancer-promoter interactions are formed, which are instructive for gene expression (Pollex et al. Nature genetics 2024).
In addition to enhancer-promoter loops, there are also many promoter-promoter (or gene-gene) chromatin loops, which often involve paralogous genes, or genes that are co-expressed (Figure 2). By making a series of careful genetic deletions, we recently showed that gene-gene loops fine-tune the coordinated expression of genes with related functions and sustain their cross-regulation (Pollex et al. Mol Cell 2024).
Currently, we are investigating how these loops are formed – what are the regulators involved and how are they controlled? To achieve this, we are integrating information from high-resolution chromatin conformation capture techniques, high-resolution imaging and genome engineering to dissect the relationship topology and transcription.
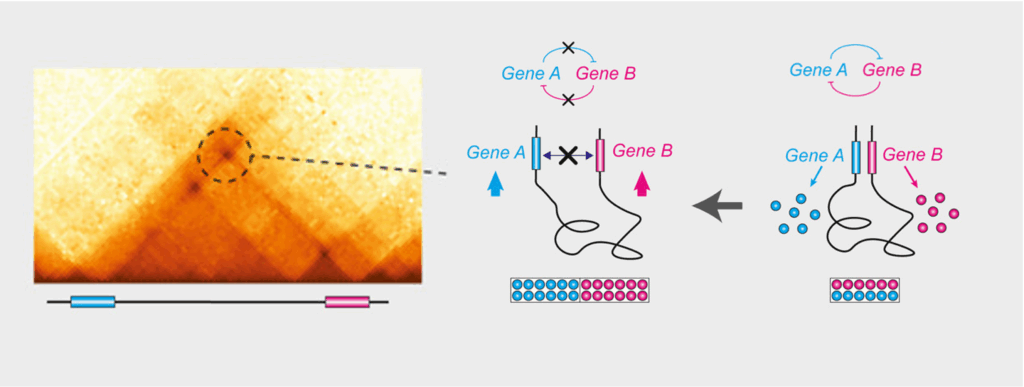
Figure 2. Gene-gene chromatin loops: 3D chromatin loops between the loci of paralogous genes act as architectural scaffolds that support their coordinated expression. Deleting either the loop connection (the loop anchor) or the genes’ promoters (maintaining the loop) alters gene expression levels and cross-regulation, revealing that the physical proximity of these genes is functionally important to fine-tune their relative levels of expression (Pollex et al. Mol Cell 2024).
Gene Regulatory Networks during Tissue & Embryonic Development
Understanding how transcription factors regulate developmental programs has been hampered in the past, as many factors regulate multiple processes at different stages of embryogenesis and even different processes in different tissues at the same stage. These pleiotropic functions often mask the role of a TF in any given tissue’s development. This is particularly problematic for TFs that are essential for the specification of a given lineage, as often those cells are not present in the mutant, and the resulting embryo can have many secondary defects, confounding the interpretation of loss-of-function studies. Single cell genomics, combined with more refined temporal perturbations, addresses many of these limitations and provides an exciting new framework to study gene regulatory networks and how they drive cell identity and differentiation. For example, we previously showed, in collaboration with Jay Shendure’s lab, that scATAC-seq, measuring open chromatin across tens of thousands of single cells, is sufficient to identify cell types, order cells along developmental trajectories, predict single cell identities, and tissue specific enhancers (Cusanovich et al. Nature 2018). We have recently scaled this up to almost 1 million cells across a time-course spanning the entirety of Drosophila embryogenesis (Calderon et al. Science 2022). This provides a comprehensive blueprint of almost all active regulatory elements across all major cell lineages during embryonic development at fine-scale temporal resolution.
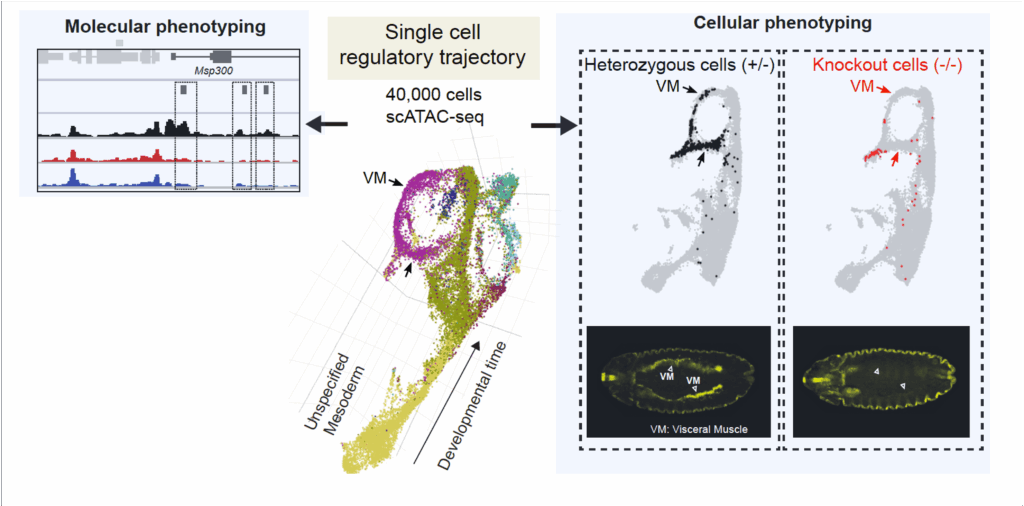
We are now combining these powerful methods with genetic perturbations using loss-of-function mutants (Secchia et al. Dev Cell 2022). This allows to phenotype developmental mutants de novo at both a cellular and molecular resolution (Figure 3). To improve the temporal resolution, we are currently using the iLEXY system to perturb TF activity for just one hour to discern their direct regulatory input.
Genetic Variation & Robustness in Developmental Programs
Variation in the non-coding genome is widely associated with quantitative traits. Many disease-associated variants are in cis-regulatory elements, yet it is very difficult to pinpoint the causal SNP within human genomes and dissect their underlying mechanism. We demonstrated that we can bridge this gap using Drosophilids, thanks to their small blocks of linkage disequilibrium (Cannavò et al. Nature 2016; Schor et al. Nature Genetics 2017). This uncovered extensive genetic epistasis within both enhancers and promoters – the functional impact of large effect variants was often buffered by the presence of a second variant that attenuates their effects.
More recently, we examined the impact of sequence variation on the binding of four essential developmental transcription factors and showed that we could use this as a fine-grained “mutagenesis” to reconstruct functionalized binding motifs. We trained a convolutional neural network to accurately predict binding from DNA sequence, which provided a mechanistic interpretation for how high effect variants impact binding (Figure 4). This uncovered unexpected relationships between these very well studied TFs, which weren’t uncovered through ‘classic’ approaches, including potential cooperative pairs and mechanisms of tissue-specific recruitment of the ubiquitous factor CTCF (Sigalova et al. Genome Research 2025 – collaboration with Judith Zaugg’s lab). Moving to the single cell level is a powerful alternative, as we showed by combining scATAC-seq with sequence variation and we developed a new framework, scDALI, to uncover cell-state-specific genetic effects (Heinen et al. Genome Biology 2022 – collaboration with Oliver Stegle’s lab).
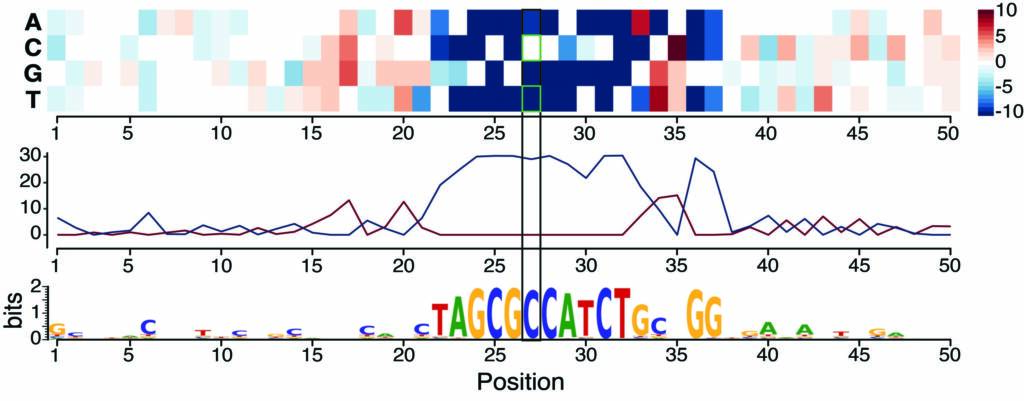
Figure 4. Training a deep neural network on ChIP data in F1 embryos prioritizes causal variants and predicts disrupted motifs: Saturation scores around one genetic variant correlate with high allelic imbalance in CTCF binding – the model predicts that the CTCF motif is disrupted by a C-to-T variant (Sigalova et al. Genome Research 2025).