Sigalova O*, Forneris M*, Stojanovska F, Zhao B, Viales RR, Rabinowitz A, Hamal F, Ballester B, Zaugg JB, Furlong EEM* These authors contributed equally to this work.Genome Research, 2025 MayRead it on: Genome Research | DOI: 10.1101/gr.279652.124
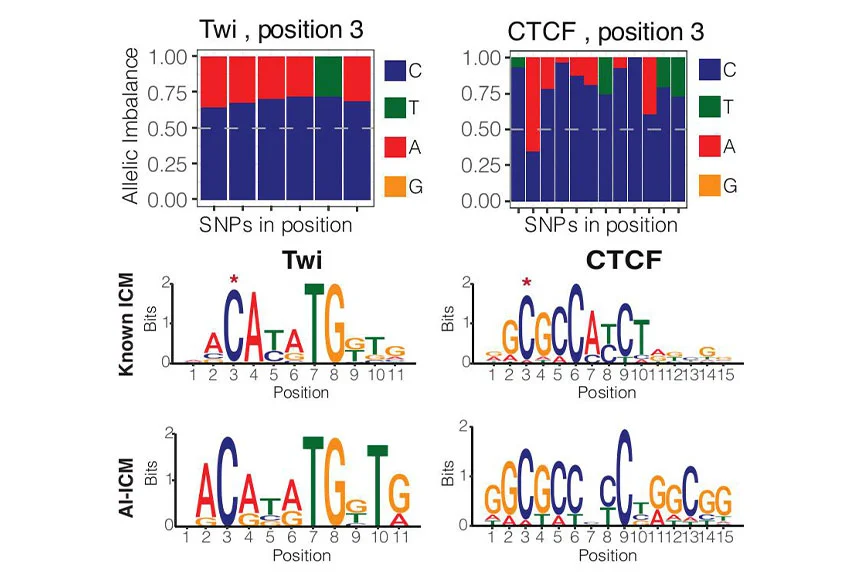
Understanding how genetic variation impacts transcription factor (TF) binding remains a major challenge, limiting our ability to model disease-associated variants. Here, we used a highly controlled system of F1 crosses with extensive genetic diversity to profile allele-specific binding of four TFs at several time points during Drosophila embryogenesis. Using a combined haplotype test, we identified 9%–18% of TF-bound regions impacted by genetic variation even for essential regulators. By expanding WASP (a tool for allele-specific read mapping) to examine indels, we increased detection of allelically imbalanced peaks by 30%–50%. This fine-grained “mutagenesis” can reconstruct functionalized binding motifs for all factors. To prioritize causal variants, we trained a convolutional neural network (Basenji) to accurately predict binding from DNA sequence. The model can also predict measured allelic imbalance for strong effect variants, providing a mechanistic interpretation for how the variant impacts binding. This reveals unexpected relationships between TFs, including potential cooperative pairs, and mechanisms of tissue-specific recruitment of the ubiquitous factor CTCF.